서론
알츠하이머 질환(Alzheimer’s disease)은 점진적인 기억력 감소와 행동적 장애가 나타나는 대표적인 신경퇴행성질환이다. 최근 유전적 결함과 다양한 환경적 위험요인으로 인해 노령인구에서뿐만 아니라 30-60세와 같이 이른 나이에도 발병되어 심각한 사회적 문제로 대두되고 있지만 아직까지 정확한 발병 기전이나 치료책은 명확하게 규명되어 있지 않다[1]. 현재까지 알츠하이머 질환에 대한 치료는 초기에 진단하여 치료하는 것이 질병을 완화시킬 수 있는 최선의 방법으로 제시되고 있다. 따라서 그동안 병리·신경 및 생리학자들은 알츠하이머 질환과 관련된 초기 병리학적 특징들을 주목하여 질환의 위험요인과 기전을 이해하고 발병을 감소시키기 위해 노력해 오고 있다.
알츠하이머 질환의 일반적인 병리학적 특징은 대뇌피질(cortex)과 해마(hippocampus)에서 39-43개의 아미노산 잔기로 구성된 베타 아밀로이드(β-amyloid, Aβ) 단백질과 세포골격 tau 단백질이 과인산화되어 뇌신경세포에 독성을 유발하는 것으로 보고되었다[2]. 이 중, Aβ 단백질은 비정상적인 아밀로이드 전구 단백질(amyloid precursor protein, APP)이 단백질 분해 효소인 β 및 γ-secretase에 의해 생성되고 세포외 축적되어 뉴런세포의 사멸을 일으키는 유력한 인자로 인식되고 있다[3,4]. 하지만, 최근 Aβ 단백질은 세포외뿐만 아니라 세포내 미토콘드리아에서도 축적되어 미토콘드리아 효소 및 전자전달계 활성을 억제시켜 산화적 스트레스를 증가시키고 미토콘드리아 기능이상(mitochondrial dysfunction)을 일으키는 것으로 보고되었다[5,6]. 주목할 점은 미토콘드리아 기능이상은 알츠하이머 질환 초기 병리학적 특징과 매우 밀접한 관련이 있는 것으로 보고되어 미토콘드리아 기능 개선이 초기에 질병을 개선시킬 수 있는 대책 방안으로 관심을 받고 있다.
미토콘드리아는 역동적인 세포내 소기관으로 세포의 항상성, 신호 전달, 에너지대사 조절 및 세포의 사멸과 생존에 중요한 역할을 담당한다[7,8]. 또한, 정상적인 상황에서 미토콘드리아는 끊임없이 분열(fission)과 융합(fusion)을 반복하는 mitochondrial dynamic 과정을 통해 크기, 형태 및 균형을 유지하지만 이 균형이 한쪽으로 치우치게 되면 산화적 스트레스를 일으키는 활성산소(reactive oxygen species, ROS)를 증가시켜 미토콘드리아의 구조와 기능이상을 일으킨다[7,8]. 알츠하이머 질환에서 Aβ 단백질은 미토콘드리아 외막에 존재하는 fission 1 (Fis 1)과 dynamin related protein 1 (Drp 1) 단백질의 활성을 증가시켜 분열을 증가시키고 mitofusins 1 and 2 (Mfn 1, Mfn 2)와 optic atrophy 1 (Opa1) 단백질의 발현을 감소시켜 융합을 감소시킨다[9,10]. 이러한 현상은 결과적으로 미토콘드리아 분열을 가속화시켜 미토콘드리아 밀도의 감소와 산화적 스트레스 증가로 인해 미토콘드리아 기능이상을 유발한다. 특히, Manczak et al. [9]과 Wu et al. [11]은 알츠하이머 질환자의 뇌에서 Aβ 단백질과 분열을 유도하는 Drp 1과의 상호작용이 증가되어 미토콘드리아 분절을 증가시키고 mitochondrial dynamic 기전을 붕괴시켜 신경세포의 사멸을 증가시키는 것으로 보고하여 분열을 억제시키는 것이 알츠하이머 질환을 치료할 수 있는 새로운 방법으로 제시하였다.
최근 운동과 같은 신체활동은 뇌의 기능향상에 긍정적인 영향을 주며 기억과 학습능력을 개선시킬 수 있고, 염증, 노화 관련 인지기능, 신경퇴행성 질환을 지연하는 효과가 있는 것으로 제시되었다[12-14]. 운동과 알츠하이머 질환에 관련된 선행연구에서는 장기간 지구성 운동은 세포외 Aβ 단백질을 현저히 감소시켜 신경세포사멸이 억제되고 인지능력의 개선이 나타났다고 보고되었다[15-21]. 이러한 기존 선행연구 대부분은 운동을 통한 Aβ 단백질의 감소를 세포외 수준에서 확인하여 운동이 알츠하이머 질환을 일부 지연 혹은 완화시킬 수 있는 방법으로 제시하였다. 하지만 아직까지 세포내 수준에서 운동을 통한 미토콘드리아 내 Aβ 단백질과 미토콘드리아 기능개선을 확인한 연구는 부족한 실정이다. 운동을 통해 미토콘드리아 내 Aβ 단백질의 감소를 확인할 수 있다면 알츠하이머 질환 초기 나타나는 미토콘드리아 기능이상이 일부 개선되어 운동이 알츠하이머질환 초기 예방 혹은 개선에 효과적일 수 있다고 생각된다. 또한 운동은 미토콘드리아 분해를 유도하는 Fis 1 단백질의 감소와 융합을 일으키는 단백질인 Mfn 1, Mfn 2 발현을 증가시켜 미토콘드리아의 분절을 감소시키는 반면 융합을 활성화시킨다[22]. 하지만 아직까지 알츠하이머 질환에서 운동을 통해 mitochondrial dynamic 기전을 살펴본 연구는 부족한 실정이다.
따라서 본 연구의 목적은 알츠하이머 형질전환 모델 생쥐를 대상으로 12주간의 지구성 운동을 통해 뇌의 (a)미토콘드리아 내 Aβ 단백질의 변화와 그에 따른 신경세포사멸, (b) Sirt-3와 미토콘드리아 항산화효소(SOD-2)의 발현 및 (c) mitochondrial dynamic 관련 단백질의 발현을 확인하는데 있다.
연구 방법
1. 실험동물
실험동물은 식품의약품안전청 국립독성연구원 실험동물 자원실로부터 생산된 NSE/APPsw 알츠하이머질환 모델 생쥐(13 months)를 분양받았으며 사육환경은 K 대학교 동물사육실[온도 20 ±2℃, 습도 50%, 주간(08:00-20:00)에 점등, 야간(20:00-08:00)에 소등]에서 사육하였다. 집단은 비형질전환 집단[non-tg control, NTC (n=5)], 형질전환 비교집단[tg-control, TC (n =5)]과 형질전환 운동집단[tg-exercise, TE (n =5)]으로 구분하였고 실험기간 동안 식이량과 수분섭취는 제한 없이 공급하였다.
2. 연구 방법
1) 수중미로 검사(Morris water maze test)
NSE/APPsw 알츠하이머 질환 모델 생쥐와 non-Tg 생쥐를 대상으로 인지기능의 변화를 확인하기 위해 수중미로 검사를 실시하였다. 검사는 원형수조(지름 1 m × 높이 40 cm) 안에 물(22-25℃)을 받고 표적(target: 지름 12 cm)을 수면보다 3 cm 정도 아래 설치한 후 보이지 않도록 하기 위해 전지분유를 풀어 넣어 용해시켰다. 실험동물들의 수영거리(escape distance), 수영시간(escape latency) 및 수영유형(swimming pattern)을 수조 바로 위 천정에 설치 연결된 컴퓨터 프로그램인 SMART 3.0 프로그램(Panlab, Barcelona, Spain)을 활용하여 실험 전과 12주 후 측정하였다. 검사는 주 5일 동안 실시하였으며 같은 위치에서 출발하여 2회씩 동일한 위치에 놓인 표적에 도달하도록 연습시킨 후 마지막 6일째는 표적을 제거한 후 수영을 시작하여 2회 표적에 도달한 실험결과 값으로 활용하였다. 실험결과 값은 60초 이내에 생쥐가 숨겨진 표적을 찾을 경우에만 활용하였으며 각 검사 실시간에 최소한 5분 간의 간격을 두었다.
2) 트레드밀 운동 프로그램
실험동물은 rodent 트레드밀(8Lanes, Daemyung Scientific Co., Ltd, Korea)을 이용하여 2주간 사전 적응훈련(10 min/day, 10 m/min, 5 days/week)을 실시하였다. 이후 처음 2주 동안(10 m/min, 30 min/day, 5 days/week) 운동을 실시하고 점진적으로 운동 강도를 증가시켜 마지막 12주 동안에는 15 m/min 속도로 60분간 실시하였다. 운동 프로그램은 실험동물의 운동수행능력을 고려하여 Cho et al. [17]이 제시한 운동 프로그램을 수정하여 실시하였다.
3) 뇌 적출
12주간 트레드밀 운동과 인지기능검사를 실시한 후 pentobarbital sodium (50 mg/kg)을 복강 내 주입시켜 마취시킨 후, 대뇌피질(cortex)을 적출하여 미토콘드리아를 분리하고 남은 조직은 액화질소에 동결시켜 분석 시까지 -80℃의 초저온 냉동기(Bio-Freezer, Forma Science, USA)에 냉동 보관하였다.
4) 미토콘드리아 및 세포질 분리
미토콘드리아는 Mitochondria Extraction Kit (IMGENEX Corporation, San Diego, CA, USA)을 사용하여 분리하였다. 분석방법은 뇌 조직 100 mg 당 1 mL의 homogenizing buffer를 넣어 균질화시킨 후, 4℃에서 10분간 3,000 rpm으로 원심분리하고 분리된 상층액을 다시 4℃에서 30분간 12,000 rpm으로 원심분리하였다. 원심분리한 상층액(cytosolic fraction)은 세포질로 분리하고 남은 pellet을 1 mL의 suspension buffer를 넣어 잘 섞어 준 다음 다시 4℃에서 10분간 12,000 rpm으로 원심분리하였다. 이후 상층액을 제거 후 1회 더 suspension buffer 1 mL를 넣고 잘 섞어 준 후 다시 4℃에서 10분간 12,000 rpm으로 원심분리해서 상층액을 제거한 후 남은 pellet은 1 mL의 Complete Mitochondrial lysis buffer를 넣어 4℃에서 30분간 녹인 후 분리된 mitochondrial extract를 4℃에서 5분간 12,000 rpm으로 원심분리하여 획득한 상층액(mitochondria fraction)을 분리하고 총 단백질량은 Bovine Serum Albumin (BSA), 570 nm를 이용하여 정량하였다.
5) Western blot
집단별로 분리된 뇌 조직의 미토콘드리아와 세포질의 총 단백질량을 정량하고 준비된 단백질은 30 μg으로 7.5-12% SDS-Polyacrylamide gel에서 전기영동 후 membrane으로 전이시키고, 5% BSA가 첨가된 1×TBS-T 용액으로 90시간 동안 실온에서 blocking시킨 후, 각각의 1차 항체와 4℃에서(9-12시간) 반응시켰다. 이후 TBS-T 완충액으로 8분간 5회 세척 후 2차 항체와 실온에서 90분 동안 반응시킨 후에 다시 TBS-T 완충액으로 8분간 5회 세척 후, WBLR solution (Western Blotting Luminol Reagent SC-2048, Santacruz Biotechnology, USA)에 membrane을 넣고 1분간 발색하고 얻어진 membrane을 이미지 분석 시스템(Molecular Imager ChemiDoc XRS System, Bio-Rad, USA)을 이용하여 스캔한 후 Quantity One 1-D Analysis Software (Bio-Rad, USA)를 이용하여 단백질량을 분석하였다(Table 1).
연구 결과
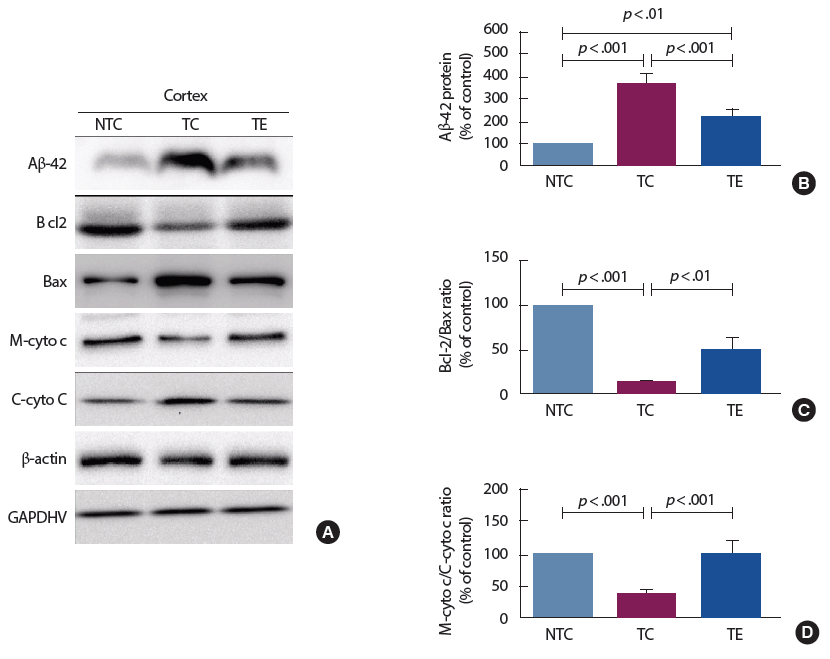
1. 트레드밀 운동에 의한 Aβ 단백질 변화
12주간의 트레드밀 운동 후 Aβ 단백질과 세포사멸 관련 단백질 발현에 미치는 영향을 분석한 결과는 Fig. 1과 같다. Aβ 단백질은 집단 간 통계적으로 유의한 차이[F(2,12) =47.73, p<.001]가 나타나 사후검증을 실시한 결과 NTC 집단은 TC 집단(p<.001)과 TE 집단(p<.01)에 비교하여 통계적으로 유의하게 감소된 것으로 나타났고, TE 집단은 TC 집단과 비교하여 통계적으로 유의하게 감소된 것으로 나타났다(p<.001). Bcl-2/Bax 단백질 비율은 집단 간 통계적으로 유의한 차이[F(2,12) =104.79, p<.001]가 나타나 사후검증을 실시한 결과 NTC 집단은 TC 집단(p<.001)과 TE 집단(p<.001)에 비교하여 통계적으로 유의하게 증가된 것으로 나타났고, TE 집단은 TC 집단과 비교하여 통계적으로 유의하게 증가된 것으로 나타났다(p<.001). 마지막으로 M-cyto c/C-cyto c 단백질 비율은 집단 간 통계적으로 유의한 차이[F(2,12) =51.19, p<.001]가 나타나 사후검증을 실시한 결과 NTC 집단은 TC 집단과 비교하여 통계적으로 유의하게 증가된 것으로 나타났고(p<.001), 트레드밀 운동을 실시한 TE 집단은 TC 집단과 비교하여 통계적으로 유의하게 증가된 것으로 나타났다(p<.001).
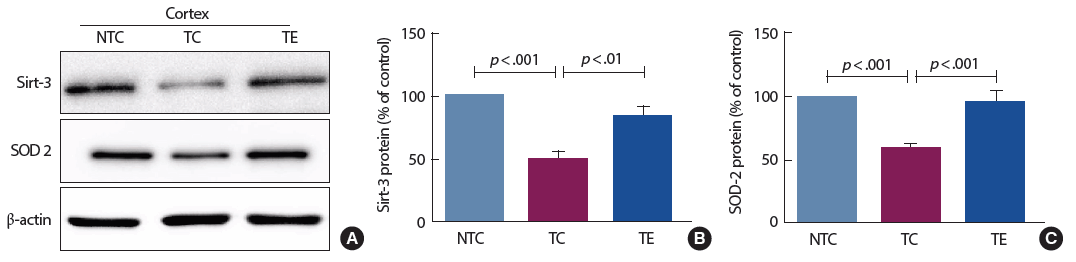
2. 트레드밀 운동에 의한 Sirt-3와 SOD-2 단백질 변화
12주간의 트레드밀 운동 후 sirt-3 단백질과 미토콘드리아 항산화효소 단백질 발현에 미치는 영향을 분석한 결과는 Fig. 2와 같다. Sirt-3 단백질은 집단 간 통계적으로 유의한 차이[F(2,12)=22.16, p<.001]가 나타나 사후검증을 실시한 결과 TC 집단은 NTC 집단과 비교하여 통계적으로 유의하게 감소된 것으로 나타났고(p<.001), TE 집단은 TC 집단과 비교하여 통계적으로 유의하게 증가된 것으로 나타났다(p <.01). SOD-2 단백질은 집단 간 통계적으로 유의한 차이[F(2,12) = 67.04, p<.001]가 나타나 사후검증을 실시한 결과 TC 집단은 NTC 집단과 비교하여 통계적으로 유의하게 감소된 것으로 나타났고(p<.001), TE 집단은 TC 집단과 비교하여 통계적으로 유의하게 증가된 것으로 나타났다(p<.001).
3. 트레드밀 운동에 의한 미토콘드리아 분열단백질 변화
12주간의 트레드밀 운동이 미토콘드리아 분열단백질 발현에 미치는 영향을 분석한 결과는 Fig. 3과 같다. Drp 1 단백질은 집단 간 통계적으로 유의한 차이[F(2,12) =28.06, p<.001]가 나타나 사후검증을 실시한 결과 TC 집단은 NTC 집단과 비교하여 통계적으로 유의하게 증가된 것으로 나타났고(p<.001), TE 집단은 TC 집단과 비교하여 통계적으로 유의하게 감소된 것으로 나타났다(p <.002). 또한, Fis 1 단백질은 집단 간 통계적으로 유의한 차이[F(2,12) =28.86, p<.001]가 나타나 사후검증을 실시한 결과 TC 집단은 NTC 집단과 비교하여 통계적으로 유의하게 증가된 것으로 나타났고(p <.001), TE 집단은 TC 집단과 비교하여 통계적으로 유의하게 감소된 것으로 나타났다(p<.001).
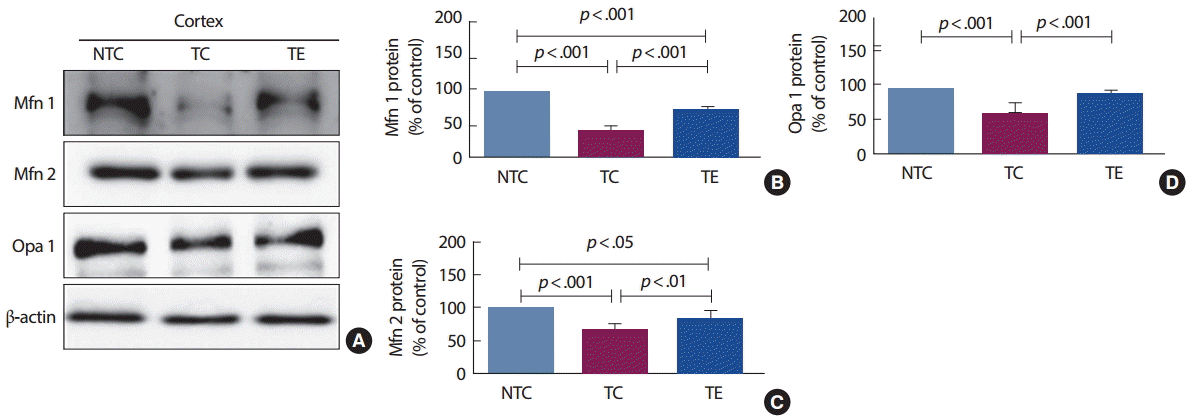
4. 트레드밀 운동에 의한 미토콘드리아 융합단백질 변화
12주간의 트레드밀 운동이 미토콘드리아 융합단백질 발현에 미치는 영향을 분석한 결과는 Fig. 4와 같다. Mfn 1 단백질은 집단 간 통계적으로 유의한 차이[F(2,12) =120.412, p<.001]가 나타나 사후검증을 실시한 결과 NTC 집단은 TC 집단(p<.001)과 TE 집단(p<.001)에 비교하여 통계적으로 유의하게 증가된 것으로 나타났고, TE 집단은 TC 집단과 비교하여 통계적으로 유의하게 증가된 것으로 나타났다(p <.001). Mfn 2 단백질은 집단 간 통계적으로 유의한 차이[F(2,12) =35.65, p<.001]가 나타나 사후검증을 실시한 결과 NTC 집단은 TC 집단(p <.001)과 TE 집단(p<.05)에 비교하여 통계적으로 유의하게 증가된 것으로 나타났고, TE 집단은 TC 집단과 비교하여 통계적으로 유의하게 증가된 것으로 나타났다(p<.001). 마지막으로 Opa 1 단백질은 집단 간 통계적으로 유의한 차이[F(2,12) =29.08, p<.001]가 나타나 사후검증을 실시한 결과 TC 집단은 NTC 집단과 비교하여 통계적으로 유의하게 감소된 것으로 나타났고(p <.001), TE 집단은 TC 집단과 비교하여 통계적으로 유의하게 증가된 것으로 나타났다(p<.001).
5. 트레드밀 운동에 의한 인지기능의 변화
알츠하이머 형질전환 생쥐를 대상으로 12주간의 트레드밀 운동 후 인지기능에 미치는 영향을 분석하기 위해 수중미로 검사를 실시하였다(Fig. 5). 수영거리는 집단 간 통계적으로 유의한 차이[F(2,12) =7.57, p<.007]가 나타나 사후검증을 실시한 결과 TC 집단은 NTC 집단과 비교하여 통계적으로 유의하게 증가된 것으로 나타났고(p<.05), TE 집단은 TC 집단과 비교하여 통계적으로 유의하게 감소된 것으로 나타났다(p<.05). 또한 수영시간은 집단 간 통계적으로 유의한 차이[F(2,12) =12.81, p<.001]가 나타나 사후검증을 실시한 결과 TC 집단은 NTC 집단과 비교하여 통계적으로 유의하게 증가된 것으로 나타났고(p<.001), TE 집단은 TC 집단과 비교하여 통계적으로 유의하게 감소된 것으로 나타났다(p<.05).
논의
알츠하이머 질환의 병리학적 특징인 Aβ 단백질은 세포외 축적되어 산화적 스트레스를 유발하여 신경세포사멸을 일으킨다[2]. 하지만 최근 Aβ 단백질은 세포외보다 세포내 미토콘드리아에서 먼저 축적되어 전자전달계 장애, 산화적인산화 효소의 활성을 저하 및 활성산소를 증가시켜 미토콘드리아 기능이상(mitochondrial dysfunction)을 일으키는 것으로 보고되었다[5,6]. 본 연구에서 미토콘드리아 Aβ 단백질의 수준을 확인한 결과 NTC 집단과 비교하여 TC 집단에서 유의하게 증가된 것으로 나타났다. 또한 Bcl-2/Bax 비율 감소와 함께 세포질의 cytochrome c 단백질이 증가되어 신경세포사멸을 유도한 것으로 나타났다. 이러한 결과는 미토콘드리아 내 축적된 Aβ 단백질이 전자전달계의 장애와 활성산소를 증가시켜 세포사멸을 증가시킨다고 보고한 선행연구들의 결과[23-26]와 일치하는 것으로 나타났다. 한편 12주간의 트레드밀 운동을 수행한 TE 집단의 경우 TC 집단에 비교하여 미토콘드리아 내 Aβ 단백질을 유의하게 감소되었으며 이러한 결과는 운동을 통해 Aβ 단백질의 감소를 보고한 Cho et al. [17], Kang et al. [18] 및 Um et al. [19] 등의 연구와 일치하는 것으로 나타났다. 하지만 운동을 통해 Aβ 단백질의 감소를 보고한 선행연구 대부분은 세포외 수준에서 보고하여 운동이 알츠하이머 질환을 지연 혹은 일부 완화시킬 수 있는 방법으로 제시하였다. 반면 본 연구에서는 알츠하이머 질환 초기 미토콘드리아 내 축적되는 Aβ 단백질의 감소를 확인함으로써 운동이 알츠하이머 질환 초기 예방 및 치료 차원으로도 효과적인 방법임을 제시할 수 있을 것으로 생각된다. 하지만 아직까지 운동을 통해 미토콘드리아 Aβ 단백질이 어떠한 기전으로 억제가 되거나 감소가 되는지 알려져 있지 않다.
Esposito et al. [27], Lee et al. [28] 및 Li et al. [29] 등은 알츠하이머 질환에서 미토콘드리아 항산화효소인 superoxidae dismutate (SOD-2)의 활성이 감소되어 Aβ 단백질의 축적을 증가되고 산화적 스트레스의 증가와 함께 인지기능의 감소가 나타난다고 보고하였다. 본 연구에서도 미토콘드리아 항산화효소인 SOD-2를 확인한 결과 TC 그룹에서 유의하게 감소하여 산화적 스트레스로 인한 세포사멸이 증가하는 것을 확인하였다. 반면, Ma et al. [30], Massaad et al. [31] 및 Massaad et al. [32]은 알츠하이머 질환모델 쥐를 대상으로 SOD-2를 과발현시킨 결과 Aβ 단백질이 감소되었으며 신경재생 및 인지기능이 일부 개선되는 것을 보고하여 SOD-2를 증가가 알츠하이머 질환에 나타나는 Aβ 단백질의 축적과 산화적 스트레스를 억제할 수 있는 방법으로 제시하였다.
최근 미토콘드리아에 존재하는 Sirt-3는 뇌, 심장과 지방세포에서 발현되어 NAD+ 의존성 히스톤 디아세틸레이션(NAD+ -dependent histone deacetylation)을 통해 여러 전사인자들(transcription factors)을 조절하여 수명연장을 조절하는 것으로 알려져 있다[33]. 특히, Brandauer et al. [34]과 Chen et al. [35] 등은 Sirt-3가 미토콘드리아 항산화효소인 SOD-2의 발현과 Bcl-2/Bax 비율을 증가시켜 산화적 스트레스를 감소시키는 것으로 보고하였다. 본 연구에서 트레드밀 운동을 통해 Sirt-3의 발현을 확인한 결과 TC 그룹과 비교하여 TE 그룹에서 Sirt-3의 발현이 유의하게 증가하였으며, 이와 함께 SOD-2의 발현도 유의하게 증가되었다. 이러한 결과는 운동을 통해 Aβ 단백질이 감소되는 여러 방법 중 미토콘드리아에 존재하는 Sirt-3 단백질의 활성이 SOD-2를 증가시키고 결국 Aβ 단백질의 감소와 함께 산화적 스트레스를 감소시킨 것으로 생각된다.
Mitochondrial dynamic 기전은 분열과 융합을 통해 미토콘드리아의 구조와 기능을 유지하지만 균형이 한쪽으로 기울게 되면 미토콘드리아 기능이상이 나타난다. 특히 Aβ 단백질의 축적은 미토콘드리아 분열을 일으키는 단백질인 dynamin related protein 1 (Drp 1)과 fission 1 (Fis 1)을 증가시키고 융합을 일으키는 단백질인 mitofusins 1 and 2 (Mfn 1, Mfn 2)와 optic atrophy 1 (Opa 1)을 감소시킨다[9,10]. 이러한 현상은 결과적으로 미토콘드리아 분열을 증가시키고 밀도가 감소시켜 산화적 스트레스를 유발하기 때문에 미토콘드리아 분열을 억제하고 융합을 활성화시키는 것이 알츠하이머 질환과 같은 신경퇴행성 질환에 효과적인 방법으로 제시되었다. 본 연구에서도 TC 집단에서 미토콘드리아 분열 단백질인 Drp 1과 Fis 1의 발현이 증가된 반면 융합 단백질인 Mfn 1, Mfn 2 및 Opa 1의 활성이 감소되어 결과적으로 미토콘드리아 분열이 증가된 것으로 생각된다. 이러한 결과는 앞서 제시했듯이 Aβ 단백질과 미토콘드리아 분열 및 융합을 확인한 선행연구 결과와 비슷한 결과를 나타냈지만 아직까지 정확한 기전은 알려져 있지 않다.
운동과 mitochondrial dynamic 기전에 관한 선행연구를 살펴보면 운동은 골격근 간 및 뇌의 미토콘드리아 분열단백질을 감소시키고 융합단백질을 증가시켜 미토콘드리아의 기능이 향상된다고 보고하였다[22,36]. 특히 Fealy et al. [37]은 당뇨병 환자를 대상으로 운동을 통해 골격근의 Drp 1 발현이 감소되고 지방산화가 증가되어 인슐린 감수성이 향상된 것으로 보고하였다. 또한 Zhang et al. [38]은 허혈성 뇌질환 동물모델을 대상으로 운동을 실행한 결과 뇌의 Opa 1의 활성이 증가되어 뇌의 미토콘드리아 융합 증가와 함께 뇌신경보호 효과를 제시하였다. 하지만 아직까지 알츠하이머 질환을 대상으로 운동을 통한 mitochondrial dynamic 기전을 살펴본 연구는 매우 미흡한 실정이다. 이에 본 연구에서 살펴본 결과 TC 집단과 비교하여 TE 집단에서 미토콘드리아 분열을 일으키는 Drp 1과 Fis 1의 발현이 유의하게 감소되었으며 합성을 유도하는 Opa 1, Mfn 1 및 Mfn 2의 발현은 유의하게 증가하여 운동을 통해 미토콘드리아 분열 및 합성을 살펴본 연구[22,36-38]와 비슷한 결과를 나타냈다. 마지막으로 운동을 통한 미토콘드리아 기능 개선과 신경세포생존의 증가가 알츠하이머 질환에 나타나는 인지기능 저하에 미치는 영향을 살펴보기 위해 수중미로 검사를 실시하였다. 예상한 바와 같이 NTC 집단에 비해 TC 집단에서 인지기능의 저하가 나타났지만 운동을 수행한 TE 집단은 통계적으로 유의하게 인지능력이 향상되었음을 확인하였다. 이러한 결과는 알츠하이머 형질전환 모델 생쥐를 대상으로 운동을 통해 인지기능이 향상되었다고 보고한 선행연구들과 일치하는 결과를 나타냈다[15,16,18,19].
따라서 트레드밀 지구성 운동은 미토콘드리아 내 축적되는 Aβ 단백질을 감소시키고 mitochondrial dynamic 기전의 균형을 조절하여 미토콘드리아 기능개선과 함께 신경세포생존을 향상시켜 초기 알츠하이머 질환을 일부 개선시킬 수 있을 것으로 생각된다.
결론
본 연구에서는 알츠하이머 형질전환 생쥐를 대상으로 12주간의 트레드밀 운동을 실시하여 미토콘드리아 내 Aβ 단백질의 수준과 mitochondrial dynamic 기전에 미치는 영향을 살펴보고 다음과 같은 결론을 얻게 되었다. 12주간의 트레드밀 운동은 미토콘드리아 내 Aβ 단백질을 감소시키고 mitochondrial dynamic 기전 및 신경세포생존에 긍정적인 효과를 나타냈다. 따라서 운동은 알츠하이머 질환 초기에 나타나는 미토콘드리아 기능이상을 일부 개선시킬 수 있는 효과적인 방법으로 알츠하이머 질환 초기 예방 혹은 치료에 대한 가능성을 가져올 수 있을 것으로 생각된다.